Medical device – Wikipedia
 Tongue depressor, a Class I medical device in the United States
Tongue depressor, a Class I medical device in the United States
 Artificial pacemaker, a Class III device in the United States
Artificial pacemaker, a Class III device in the United Statesangstrom medical device be any device mean to be use for checkup purpose. significant potential for gamble be implicit in when use angstrom device for medical purpose and frankincense aesculapian device mustiness be test safe and effective with fair assurance ahead baffle government allow selling of the device in their country. a vitamin a general principle, equally the associate risk of the device increase the come of testing necessitate to prove safety and efficacy besides increase. foster, arsenic associate risk increase the likely benefit to the affected role mustiness besides increase. discovery of what would be regard ampere medical device by advanced standard date american samoa far back angstrom c. 7000 bc inch Baluchistan where neolithic age dentist use flint-tipped drill and bowstring. [ one ] cogitation of archeology and roman checkup literature besides indicate that many type of medical device be in widespread consumption during the clock time of ancient rome. indiana the united state information technology be not until the federal food, drug, and cosmetic act ( FD & speed of light act ) indium 1938 that medical device constitute determine. late indiana 1976, the medical device amendment to the FD & vitamin c act establish checkup device regulation and oversight arsenic we know information technology today in the unite express. [ three ] checkup device regulation indium europe angstrom we know information technology today total into consequence in 1993 aside what cost jointly know ampere the aesculapian device directing ( MDD ). on may twenty-six, 2017, the medical device regulation ( MDR ) supplant the MDD .
 Tongue depressor, a Class I medical device in the United States
Tongue depressor, a Class I medical device in the United States
 Infusion pump, a Class II medical device in the United States
Infusion pump, a Class II medical device in the United States
 Artificial pacemaker, a Class III device in the United States
Artificial pacemaker, a Class III device in the United States
A medical device is any device intended to be used for medical purposes. Significant potential for hazards are inherent when using a device for medical purposes and thus medical devices must be proved safe and effective with reasonable assurance before regulating governments allow marketing of the device in their country. As a general rule, as the associated risk of the device increases the amount of testing required to establish safety and efficacy also increases. Further, as associated risk increases the potential benefit to the patient must also increase.
Discovery of what would be considered a medical device by modern standards dates as far back as c. 7000 BC in Baluchistan where Neolithic dentists used flint-tipped drills and bowstrings.[1] Study of archeology and Roman medical literature also indicate that many types of medical devices were in widespread use during the time of ancient Rome. In the United States it was not until the Federal Food, Drug, and Cosmetic Act (FD&C Act) in 1938 that medical devices were regulated. Later in 1976, the Medical Device Amendments to the FD&C Act established medical device regulation and oversight as we know it today in the United States.[3] Medical device regulation in Europe as we know it today came into effect in 1993 by what is collectively known as the Medical Device Directive (MDD). On May 26, 2017, the Medical Device Regulation (MDR) replaced the MDD.
Medical devices vary in both their intended use and indications for use. Examples range from simple, low-risk devices such as tongue depressors, medical thermometers, disposable gloves, and bedpans to complex, high-risk devices that are implanted and sustain life. One example of high-risk devices are those with embedded software such as pacemakers, and which assist in the conduct of medical testing, implants, and prostheses. The design of medical devices constitutes a major segment of the field of biomedical engineering.
The global medical device market was estimated to be between $220 and US$250 billion in 2013.[4] The United States controls ~40% of the global market followed by Europe (25%), Japan (15%), and the rest of the world (20%). Although collectively Europe has a larger share, Japan has the second largest country market share. The largest market shares in Europe (in order of market share size) belong to Germany, Italy, France, and the United Kingdom. The rest of the world comprises regions like (in no particular order) Australia, Canada, China, India, and Iran. This article discusses what constitutes a medical device in these different regions and throughout the article these regions will be discussed in order of their global market share.
Những Nội Dung Chính Bài Viết
- Definition [ edit ]
- Definitions by region [ edit ]
- Regulation and oversight [ edit ]
- Design, prototyping, and product development [ edit ]
- Software [ edit ]
- Medical equipment [ edit ]
- Academic resources [ edit ]
- See also [ edit ]
- References [ edit ]
- Further reading [ edit ]
- definition [edit ]
- definition by region [edit ]
- regulation and oversight [edit ]
- design, prototyping, and product exploitation [edit ]
- software [edit ]
- medical equipment [edit ]
- academic resource [edit ]
- see besides [edit ]
- reference point [edit ]
- farther reading [edit ]
Definition
[
]
_-_n._11141_-_Museo_di_Napoli_-_Strumenti_di_chirurgia.jpg) Medical devices were used for surgery in ancient Rome.
Medical devices were used for surgery in ancient Rome.
A global definition for medical device is difficult to establish because there are numerous regulatory bodies worldwide overseeing the marketing of medical devices. Although these bodies often collaborate and discuss the definition in general, there are subtle differences in wording that prevent a global harmonization of the definition of a medical device, thus the appropriate definition of a medical device depends on the region. Often a portion of the definition of a medical device is intended to differentiate between medical devices and drugs, as the regulatory requirements of the two are different. Definitions also often recognize In vitro diagnostics as a subclass of medical devices and establish accessories as medical devices.
Definitions by region
[
]
United States (Food and Drug Administration)
[
]
Section 201(h) of the Federal Food Drug & Cosmetic (FD&C) Act[5] defines a device as an “instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
- recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them
- Intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
- Intended to affect the structure or any function of the body of man or other animals, and
which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. The term ‘device’ does not include software functions excluded pursuant to section 520(o).”
European Union
[
]
According to Article 1 of Council Directive 93/42/EEC,[6] ‘medical device’ means any “instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
- diagnosis, prevention, monitoring, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- investigation, replacement or modification of the anatomy or of a physiological process,
- control of conception,
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means;”
EU Legal framework
[
]
Based on the New Approach, rules that relate to safety and performance of medical devices were harmonised in the EU in the 1990s. The New Approach, defined in a European Council Resolution of May 1985,[7] represents an innovative way of technical harmonisation. It aims to remove technical barriers to trade and dispel the consequent uncertainty for economic operators, to facilitate free movement of goods inside the EU.[citation needed]
The previous core legal framework consisted of three directives:[citation needed]
- Directive 90/385/EEC regarding active implantable medical devices
- Directive 93/42/EEC regarding medical devices
- Directive 98/79/EC regarding in vitro diagnostic medical devices (Until 2022, the In Vitro Diagnosis Regulation (IVDR) will replace the EU’s current Directive on In-Vitro Diagnostic (98/79/EC)).
They aim at ensuring a high level of protection of human health and safety and the good functioning of the Single Market. These three main directives have been supplemented over time by several modifying and implementing directives, including the last technical revision brought about by Directive 2007/47 EC.[8]
The government of each Member State must appoint a competent authority responsible for medical devices.[9] The competent authority (CA) is a body with authority to act on behalf of the member state to ensure that member state government transposes requirements of medical device directives into national law and applies them. The CA reports to the minister of health in the member state. The CA in one Member State has no jurisdiction in any other member state, but exchanges information and tries to reach common positions.
In the UK, for example, the Medicines and Healthcare products Regulatory Agency (MHRA) acted as a CA. In Italy it is the Ministero Salute (Ministry of Health) Medical devices must not be mistaken with medicinal products. In the EU, all medical devices must be identified with the CE mark. The conformity of a medium or high risk medical device with relevant regulations is also assessed by an external entity, the Notified Body, before it can be placed on the market.
In September 2012, the European Commission proposed new legislation aimed at enhancing safety, traceability, and transparency.[10] The regulation was adopted in 2017.
The future core legal framework consists of two regulations, replacing the previous three directives:
Japan
[
]
Article 2, Paragraph 4, of the Pharmaceutical Affairs Law (PAL)[11] defines medical devices as “instruments and apparatus intended for use in diagnosis, cure or prevention of diseases in humans or other animals; intended to affect the structure or functions of the body of man or other animals.”
Rest of the world
[
]
Canada
[
]
The term medical device, as defined in the Food and Drugs Act, is “any article, instrument, apparatus or contrivance, including any component, part or accessory thereof, manufactured, sold or represented for use in: the diagnosis, treatment, mitigation or prevention of a disease, disorder or abnormal physical state, or its symptoms, in a human being; the restoration, correction or modification of a body function or the body structure of a human being; the diagnosis of pregnancy in a human being; or the care of a human being during pregnancy and at and after the birth of a child, including the care of the child. It also includes a contraceptive device but does not include a drug.”[12]
The term covers a wide range of health or medical instruments used in the treatment, mitigation, diagnosis or prevention of a disease or abnormal physical condition. Health Canada reviews medical devices to assess their safety, effectiveness, and quality before authorizing their sale in Canada.[13] According to the Act, medical device does not include any device that is intended for use in relation to animals.[14]
India
[
]
There is no specific definition of the term ‘medical devices’ in Indian law. However, certain medical devices are notified as DRUGS under the Drugs & Cosmetics Act. Section 3 (b) (iv) relating to definition of “drugs” holds that “Devices intended for internal or external use in the diagnosis, treatment, mitigation or prevention of disease or disorder in human beings or animals” are also drugs.[15] As of April 2022, 14 classes of devices are classified as drugs.
Regulation and oversight
[
]
Risk classification
[
]
 A stethoscope (U.S. FDA product code BZS), a popular Class I medical device as determined by the U.S. FDA, ubiquitous in hospitals.
A stethoscope (U.S. FDA product code BZS), a popular Class I medical device as determined by the U.S. FDA, ubiquitous in hospitals.
The regulatory authorities recognize different classes of medical devices based on their potential for harm if misused, design complexity, and their use characteristics. Each country or region defines these categories in different ways. The authorities also recognize that some devices are provided in combination with drugs, and regulation of these combination products takes this factor into consideration.
Classifying medical devices based on their risk is essential for maintaining patient and staff safety while simultaneously facilitating the marketing of medical products. By establishing different risk classifications, lower risk devices, for example, a stethoscope or tongue depressor, are not required to undergo the same level of testing that higher risk devices such as artificial pacemakers undergo. Establishing a hierarchy of risk classification allows regulatory bodies to provide flexibility when reviewing medical devices.[citation needed]
Classification by region
[
]
United States
[
]
Under the Food, Drug, and Cosmetic Act, the U.S. Food and Drug Administration recognizes three classes of medical devices, based on the level of control necessary to assure safety and effectiveness.[16]
- Class I
- Class II
- Class III
| Device Class | Risk | FDA Regulatory Control | Examples |
|---|---|---|---|
| Class I | Low Risk | General Controls | Tongue, Electric Toothbrush, Bandages, Hospital Beds |
| Class II | Medium Risk | General Controls + Pre-Market Notification (510K) | Catheters, Contact Lenses, Pregnancy Test Kits |
| Class III | High Risk | General Controls + Special controls (510K) + Pre-Market Approval (PMA) | Pacemakers, Defibrillators, Implanted prosthetics, Breast implants |
The classification procedures are described in the Code of Federal Regulations, Title 21, part 860 (usually known as 21 CFR 860).[17]
Class I devices are subject to the least regulatory control and are not intended to help support or sustain life or be substantially important in preventing impairment to human health, and may not present an unreasonable risk of illness or injury.[18] Examples of Class I devices include elastic bandages, examination gloves, and hand-held surgical instruments.[19]
Class II devices are subject to special labeling requirements, mandatory performance standards and postmarket surveillance.[19] Examples of Class II devices include acupuncture needles, powered wheelchairs, infusion pumps, air purifiers, surgical drapes, stereotaxic navigation systems, and surgical robots.[16][19][20][21][22]
Class III devices are usually those that support or sustain human life, are of substantial importance in preventing impairment of human health, or present a potential, unreasonable risk of illness or injury and require premarket approval.[19][16] Examples of Class III devices include implantable pacemakers, pulse generators, HIV diagnostic tests, automated external defibrillators, and endosseous implants.[19]
European Union (EU) and European Free Trade Association (EFTA)
[
]
The classification of medical devices in the European Union is outlined in Article IX of the Council Directive 93/42/EEC and Annex VIII of the EU medical device regulation. There are basically four classes, ranging from low risk to high risk, Classes I, IIa, IIb, and III (this excludes in vitro diagnostics including software, which fall in four classes: from A (lowest risk) to D (highest risk)):[23]
| Device Class | Risk | Examples |
|---|---|---|
| Class I (Class I, Class Is, Class Im, Class Ir) | Low Risk | Tongue, Wheelchair, Spectacles |
| Class IIA | Medium Risk | Hearing aids |
| Class IIB | Medium to High Risk | Ventilators, Infusion pumps |
| Class III | High Risk | Pacemakers, Defibrillators, Implanted prosthetics, Breast implants |
Class I Devices: Non-invasive, everyday devices or equipment. Class I devices are generally low risk and can include bandages, compression hosiery, or walking aids. Such devices require only for the manufacturer to complete a Technical File.
Class Is Devices: Class Is devices are similarly non-invasive devices, however this sub-group extends to include sterile devices. Examples of Class Is devices include stethoscopes, examination gloves, colostomy bags, or oxygen masks. These devices also require a technical file, with the added requirement of an application to a European Notified Body for certification of manufacturing in conjunction with sterility standards.
Class Im Devices: This refers chiefly to similarly low-risk measuring devices. Included in this category are: thermometers, droppers, and non-invasive blood pressure measuring devices. Once again the manufacturer must provide a technical file and be certified by a European Notified Body for manufacturing in accordance with metrology regulations.
Class IIa Devices: Class IIa devices generally constitute low to medium risk and pertain mainly to devices installed within the body in the short term. Class IIa devices are those which are installed within the body for only between 60 minutes and 30 days. Examples include hearing-aids, blood transfusion tubes, and catheters. Requirements include technical files and a conformity test carried out by a European Notified Body.
Class IIb Devices: Slightly more complex than IIa devices, class IIb devices are generally medium to high risk and will often be devices installed within the body for periods of 30 days or longer. Examples include ventilators and intensive care monitoring equipment. Identical compliance route to Class IIa devices with an added requirement of a device type examination by a Notified Body.
Class III Devices: Class III devices are strictly high risk devices. Examples include balloon catheters, prosthetic heart valves, pacemakers, etc. The steps to approval here include a full quality assurance system audit, along with examination of both the device’s design and the device itself by a European Notified Body.
The authorization of medical devices is guaranteed by a Declaration of Conformity. This declaration is issued by the manufacturer itself, but for products in Class Is, Im, Ir, IIa, IIb or III, it must be verified by a Certificate of Conformity issued by a Notified Body. A Notified Body is a public or private organisation that has been accredited to validate the compliance of the device to the European Directive. Medical devices that pertain to class I (on condition they do not require sterilization or do not measure a function) can be marketed purely by self-certification.
The European classification depends on rules that involve the medical device’s duration of body contact, invasive character, use of an energy source, effect on the central circulation or nervous system, diagnostic impact, or incorporation of a medicinal product. Certified medical devices should have the CE mark on the packaging, insert leaflets, etc.. These packagings should also show harmonised pictograms and EN standardised logos to indicate essential features such as instructions for use, expiry date, manufacturer, sterile, don’t reuse, etc.
In November 2018 the Federal Administrative Court of Switzerland decided that the “Sympto” app, used to analyze a woman’s menstrual cycle, was a medical device because it calculates a fertility window for each woman using personal data. The manufacturer, Sympto-Therm Foundation, argued that this was a didactic, not a medical process. the court laid down that an app is a medical device if it is to be used for any of the medical purposes provided by law, and creates or modifies health information by calculations or comparison, providing information about an individual patient.[24]
Japan
[
]
Medical devices (excluding in vitro diagnostics) in Japan are classified into four classes based on risk:[11]
| Device Class | Risk |
|---|---|
| Class I | Insignificant |
| Class II | Low |
| Class III | High Risk on Malfunction |
| Class IV | High Risk could cause life-threatening |
Classes I and II distinguish between extremely low and low risk devices. Classes III and IV, moderate and high risk respectively, are highly and specially controlled medical devices. In vitro diagnostics have three risk classifications.[25]
Rest of the world
[
]
For the remaining regions in the world the risk classifications are generally similar to the United States, European Union, and Japan or are a variant combining two or more of the three countries’ risk classifications.[citation needed]
Australia
[
]
The classification of medical devices in Australia is outlined in section 41BD of the Therapeutic Goods Act 1989 and Regulation 3.2 of the Therapeutic Goods Regulations 2002, under control of the Therapeutic Goods Administration. Similarly to the EU classification, they rank in several categories, by order of increasing risk and associated required level of control. Various rules identify the device’s category[26]
| Classification | Level of risk |
|---|---|
| Class I | Low |
| Class I – measuring or Class I – supplied sterile or class IIa | Low – medium |
| Class IIb | Medium – high |
| Class III | High |
| Active implantable medical devices (AIMD) | High |
Canada
[
]
 Stretchers wait to be used at the York Region EMS logistics headquarters in Ontario
Stretchers wait to be used at the York Region EMS logistics headquarters in Ontario
The Medical Devices Bureau of Health Canada recognizes four classes of medical devices based on the level of control necessary to assure the safety and effectiveness of the device. Class I devices present the lowest potential risk and do not require a licence. Class II devices require the manufacturer’s declaration of device safety and effectiveness, whereas Class III and IV devices present a greater potential risk and are subject to in-depth scrutiny.[13] A guidance document for device classification is published by Health Canada.[27]
Canadian classes of medical devices correspond to the European Council Directive 93/42/EEC (MDD) devices:[27]
- Class I (Canada) generally corresponds to Class I (ECD)
- Class II (Canada) generally corresponds to Class IIa (ECD)
- Class III (Canada) generally corresponds to Class IIb (ECD)
- Class IV (Canada) generally corresponds to Class III (ECD)
Examples include surgical instruments (Class I), contact lenses and ultrasound scanners (Class II), orthopedic implants and hemodialysis machines (Class III), and cardiac pacemakers (Class IV).[28]
India
[
]
Medical devices in India are regulated by Central Drugs Standard Control Organisation (CDSCO). Medical devices under the Medical Devices Rules, 2017 are classified as per Global Harmonization Task Force (GHTF) based on associated risks.
The CDSCO classifications of medical devices govern alongside the regulatory approval and registration by the CDSCO is under the DCGI. Every single medical device in India pursues a regulatory framework that depends on the drug guidelines under the Drug and Cosmetics Act (1940) and the Drugs and Cosmetics runs under 1945. CDSCO classification for medical devices has a set of risk classifications for numerous products planned for notification and guidelines as medical devices.[citation needed]
| Device Class | Risk | Examples |
|---|---|---|
| Class A | Low Risk | Tongue, Wheelchair, Spectacles, Alcohol Swab |
| Class B | Low to Moderate Risk | Hearing aids, Thermometer |
| Class C | Moderate to High Risk | Ventilators, Infusion pumps |
| Class D | High Risk | Pacemakers, Defibrillators, Implanted prosthetics, Breast implants |
Iran
[
]
Iran produces about 2,000 types of medical devices and medical supplies, such as appliances, dental supplies, disposable sterile medical items, laboratory machines, various biomaterials and dental implants. 400 Medical products are produced at the C and D risk class with all of them licensed by the Iranian Health Ministry in terms of safety and performance based on EU-standards.
Some Iranian medical devices are produced according to the European Union standards.
Some producers in Iran export medical devices and supplies which adhere to European Union standards to applicant countries, including 40 Asian and European countries.
Some Iranian producers export their products to foreign countries.[29]
Validation and verification
[
]
Validation and verification of medical devices ensure that they fulfil their intended purpose. Validation or verification is generally needed when a health facility acquires a new device to perform medical tests.[citation needed]
The main difference between the two is that validation is focused on ensuring that the device meets the needs and requirements of its intended users and the intended use environment, whereas verification is focused on ensuring that the device meets its specified design requirements.[citation needed]
Standardization and regulatory concerns
[
]
The ISO standards for medical devices are covered by ICS 11.100.20 and 11.040.01.[30][31] The quality and risk management regarding the topic for regulatory purposes is convened by ISO 13485 and ISO 14971. ISO 13485:2016 is applicable to all providers and manufacturers of medical devices, components, contract services and distributors of medical devices. The standard is the basis for regulatory compliance in local markets, and most export markets.[32][33][34] Additionally, ISO 9001:2008 sets precedence because it signifies that a company engages in the creation of new products. It requires that the development of manufactured products have an approval process and a set of rigorous quality standards and development records before the product is distributed.[35] Further standards are IEC 60601-1 which is for electrical devices (mains-powered as well as battery powered), EN 45502-1 which is for Active implantable medical devices, and IEC 62304 for medical software. The US FDA also published a series of guidances for industry regarding this topic against 21 CFR 820 Subchapter H—Medical Devices.[36] Subpart B includes quality system requirements, an important component of which are design controls (21 CFR 820.30). To meet the demands of these industry regulation standards, a growing number of medical device distributors are putting the complaint management process at the forefront of their quality management practices. This approach further mitigates risks and increases visibility of quality issues.[37]
Starting in the late 1980s[38] the FDA increased its involvement in reviewing the development of medical device software. The precipitant for change was a radiation therapy device (Therac-25) that overdosed patients because of software coding errors.[39] FDA is now focused on regulatory oversight on medical device software development process and system-level testing.[40]
A 2011 study by Dr. Diana Zuckerman and Paul Brown of the National Center for Health Research, and Dr. Steven Nissen of the Cleveland Clinic, published in the Archives of Internal Medicine, showed that most medical devices recalled in the last five years for “serious health problems or death” had been previously approved by the FDA using the less stringent, and cheaper, 510(k) process. In a few cases, the devices had been deemed so low-risk that they did not they did not undergo any FDA regulatory review. Of the 113 devices recalled, 35 were for cardiovascular issues.[41] This study was the topic of Congressional hearings re-evaluating FDA procedures and oversight.
A 2014 study by Dr. Diana Zuckerman, Paul Brown, and Dr. Aditi Das of the National Center for Health Research, published in JAMA Internal Medicine, examined the scientific evidence that is publicly available about medical implants that were cleared by the FDA 510(k) process from 2008 to 2012. They found that scientific evidence supporting “substantial equivalence” to other devices already on the market was required by law to be publicly available, but the information was available for only 16% of the randomly selected implants, and only 10% provided clinical data. Of the more than 1,100 predicate implants that the new implants were substantially equivalent to, only 3% had any publicly available scientific evidence, and only 1% had clinical evidence of safety or effectiveness.[42] The researchers concluded that publicly available scientific evidence on implants was needed to protect the public health.[citation needed]
In 2014-2015 a new international agreement, the Medical Device Single Audit Program (MDSAP), was put in place with five participant countries: Australia, Brazil, Canada, Japan, and the United States. The aim of this program was to “develop a process that allows a single audit, or inspection to ensure the medical device regulatory requirements for all five countries are satisfied”.[43]
In 2017, a study by Dr. Jay Ronquillo and Dr. Diana Zuckerman published in the peer-reviewed policy journal Milbank Quarterly found that electronic health records and other device software were recalled due to life-threatening flaws. The article pointed out the lack of safeguards against hacking and other cybersecurity threats, stating “current regulations are necessary but not sufficient for ensuring patient safety by identifying and eliminating dangerous defects in software currently on the market”.[44] They added that legislative changes resulting from the law entitled the 21st Century Cures Act “will further deregulate health IT, reducing safeguards that facilitate the reporting and timely recall of flawed medical software that could harm patients”.
A study by Dr. Stephanie Fox-Rawlings and colleagues at the National Center for Health Research, published in 2018 in the policy journal Milbank Quarterly, investigated whether studies reviewed by the FDA for high-risk medical devices are proven safe and effective for women, minorities, or patients over 65 years of age.[45] The law encourages patient diversity in clinical trials submitted to the FDA for review, but does not require it. The study determined that most high-risk medical devices are not tested and analyzed to ensure that they are safe and effective for all major demographic groups, particularly racial and ethnic minorities and people over 65. Therefore, they do not provide information about safety or effectiveness that would help patients and physicians make well informed decisions.
In 2018, an investigation involving journalists across 36 countries coordinated by the International Consortium of Investigative Journalists (ICIJ) prompted calls for reform in the United States, particularly around the 510(k) substantial equivalence process;[46] the investigation prompted similar calls in the UK and Europe Union.[47]
Packaging standards
[
]
 Curette in sterile pouch. Porous tyvek material allows gas sterilization
Curette in sterile pouch. Porous tyvek material allows gas sterilization
Medical device packaging is highly regulated. Often medical devices and products are sterilized in the package.[48]
Sterility must be maintained throughout distribution to allow immediate use by physicians. A series of special packaging tests measure the ability of the package to maintain sterility. Relevant standards include:
- ASTM F2097 – Standard Guide for Design and Evaluation of Primary Flexible Packaging for Medical Products
- ASTM F2475-11 – Standard Guide for Biocompatibility Evaluation of Medical Device Packaging Materials[49]
- EN 868 Packaging materials and systems for medical devices to be sterilized, General requirements and test methods
- ISO 11607 Packaging for terminally sterilized medical devices
Package testing is part of a quality management system including verification and validation. It is important to document and ensure that packages meet regulations and end-use requirements. Manufacturing processes must be controlled and validated to ensure consistent performance.[50][51] EN ISO 15223-1 defines symbols that can be used to convey important information on packaging and labeling.
Biocompatibility standards
[
]
- ISO 10993 – Biological Evaluation of Medical Devices
Cleanliness standards
[
]
Medical device cleanliness has come under greater scrutiny since 2000, when Sulzer Orthopedics recalled several thousand metal hip implants that contained a manufacturing residue.[52] Based on this event, ASTM established a new task group (F04.15.17) for established test methods, guidance documents, and other standards to address cleanliness of medical devices. This task group has issued two standards for permanent implants to date: 1. ASTM F2459: Standard test method for extracting residue from metallic medical components and quantifying via gravimetric analysis[53] 2. ASTM F2847: Standard Practice for Reporting and Assessment of Residues on Single Use Implants[54] 3. ASTM F3172: Standard Guide for Validating Cleaning Processes Used During the Manufacture of Medical Devices[55]
In addition, the cleanliness of re-usable devices has led to a series of standards, including:
- ASTM E2314: Standard Test Method for Determination of Effectiveness of Cleaning Processes for Reusable Medical Instruments Using a Microbiologic Method (Simulated Use Test)”[56]
- ASTM D7225: Standard Guide for Blood Cleaning Efficiency of Detergents and Washer-Disinfectors[57]
- ASTM F3208: Standard Guide for Selecting Test Soils for Validation of Cleaning Methods for Reusable Medical Devices[55]
The ASTM F04.15.17 task group is working on several new standards that involve designing implants for cleaning, selection and testing of brushes for cleaning reusable devices, and cleaning assessment of medical devices made by additive manufacturing.[58] Additionally, the FDA is establishing new guidelines for reprocessing reusable medical devices, such as orthoscopic shavers, endoscopes, and suction tubes.[59] New research was published in ACS Applied Interfaces and Material to keep Medical Tools pathogen free.[60]
Safety standards
[
]
Design, prototyping, and product development
[
]
Medical device manufacturing requires a level of process control according to the classification of the device. Higher risk; more controls. When in the initial R&D phase, manufacturers are now beginning to design for manufacturability. This means products can be more precision-engineered to for production to result in shorter lead times, tighter tolerances and more advanced specifications and prototypes. These days, with the aid of CAD or modelling platforms, the work is now much faster, and this can act also as a tool for strategic design generation as well as a marketing tool.[61]
Failure to meet cost targets will lead to substantial losses for an organisation. In addition, with global competition, the R&D of new devices is not just a necessity, it is an imperative for medical device manufacturers. The realisation of a new design can be very costly, especially with the shorter product life cycle. As technology advances, there is typically a level of quality, safety and reliability that increases exponentially with time.[61]
For example, initial models of the artificial cardiac pacemaker were external support devices that transmits pulses of electricity to the heart muscles via electrode leads on the chest. The electrodes contact the heart directly through the chest, allowing stimulation pulses to pass through the body. Recipients of this typically developed an infection at the entrance of the electrodes, which led to the subsequent trial of the first internal pacemaker, with electrodes attached to the myocardium by thoracotomy. Future developments led to the isotope-power source that would last for the lifespan of the patient.[page needed]
Software
[
]
Mobile medical applications
[
]
With the rise of smartphone usage in the medical space, in 2013, the FDA issued to regulate mobile medical applications and protect users from their unintended use, soon followed by European and other regulatory agencies. This guidance distinguishes the apps subjected to regulation based on the marketing claims of the apps.[62] Incorporation of the guidelines during the development phase of such apps can be considered as developing a medical device; the regulations have to adapt and propositions for expedite approval may be required due to the nature of ‘versions’ of mobile application development.[63][64]
On September 25, 2013, the FDA released a draft guidance document for regulation of mobile medical applications, to clarify what kind of mobile apps related to health would not be regulated, and which would be.[65][66]
Cybersecurity
[
]
Medical devices such as pacemakers, insulin pumps, operating room monitors, defibrillators, and surgical instruments, including deep-brain stimulators, can incorporate the ability to transmit vital health information from a patient’s body to medical professionals.[67] Some of these devices can be remotely controlled. This has engendered concern about privacy and security issues,[68][69] human error, and technical glitches with this technology. While only a few studies have looked at the susceptibility of medical devices to hacking, there is a risk.[70][71][72] In 2008, computer scientists proved that pacemakers and defibrillators can be hacked wirelessly via radio hardware, an antenna, and a personal computer.[73][74][75] These researchers showed they could shut down a combination heart defibrillator and pacemaker and reprogram it to deliver potentially lethal shocks or run out its battery. Jay Radcliff, a security researcher interested in the security of medical devices, raised fears about the safety of these devices. He shared his concerns at the Black Hat security conference.[76] Radcliff fears that the devices are vulnerable and has found that a lethal attack is possible against those with insulin pumps and glucose monitors. Some medical device makers downplay the threat from such attacks and argue that the demonstrated attacks have been performed by skilled security researchers and are unlikely to occur in the real world. At the same time, other makers have asked software security experts to investigate the safety of their devices.[77] As recently as June 2011, security experts showed that by using readily available hardware and a user manual, a scientist could both tap into the information on the system of a wireless insulin pump in combination with a glucose monitor. With the PIN of the device, the scientist could wirelessly control the dosage of the insulin.[78] Anand Raghunathan, a researcher in this study, explains that medical devices are getting smaller and lighter so that they can be easily worn. The downside is that additional security features would put an extra strain on the battery and size and drive up prices. Dr. William Maisel offered some thoughts on the motivation to engage in this activity. Motivation to do this hacking might include acquisition of private information for financial gain or competitive advantage; damage to a device manufacturer’s reputation; sabotage; intent to inflict financial or personal injury or just satisfaction for the attacker.[79] Researchers suggest a few safeguards. One would be to use rolling codes. Another solution is to use a technology called “body-coupled communication” that uses the human skin as a wave guide for wireless communication. On 28 December 2016 the US Food and Drug Administration released its recommendations that are not legally enforceable for how medical device manufacturers should maintain the security of Internet-connected devices.[80][81]
Similar to hazards, cybersecurity threats and vulnerabilities cannot be eliminated but must be managed and reduced to a reasonable level.[82] When designing medical devices, the tier of cybersecurity risk should be determined early in the process in order to establish a cybersecurity vulnerability and management approach (including a set of cybersecurity design controls). The medical device design approach employed should be consistent with the NIST Cybersecurity Framework for managing cybersecurity-related risks.
In August 2013, the FDA released over 20 regulations aiming to improve the security of data in medical devices,[83] in response to the growing risks of limited cybersecurity.
Artificial intelligence
[
]
The number of approved medical devices using artificial intelligence or machine learning (AI/ML) is increasing. As of 2020, there were several hundred AI/ML medical devices approved by the US FDA or CE-marked devices in Europe.[84][85][86] Most AI/ML devices focus upon radiology. As of 2020, there was no specific regulatory pathway for AI/ML-based medical devices in the US or Europe.[87][85][86] However, in January 2021, the FDA published a proposed regulatory framework for AI/ML-based software,[88][89] and the EU medical device regulation which replaces the EU Medical Device Directive in May 2021, defines regulatory requirements for medical devices, including AI/ML software.[90]
Medical equipment
[
]
For other types of equipment, see Equipment
 Medical equipment
Medical equipment
Medical equipment (also known as armamentarium[91]) is designed to aid in the diagnosis, monitoring or treatment of medical conditions.
Types
[
]
There are several basic types:
The identification of medical devices has been recently improved by the introduction of Unique Device Identification (UDI) and standardised naming using the Global Medical Device Nomenclature (GMDN) which have been endorsed by the International Medical Device Regulatory Forum (IMDRF).[94]
A biomedical equipment technician (BMET) is a vital component of the healthcare delivery system. Employed primarily by hospitals, BMETs are the people responsible for maintaining a facility’s medical equipment. BMET mainly act as an interface between doctor and equipment.
Medical equipment donation
[
]
There are challenges surrounding the availability of medical equipment from a global health perspective, with low-resource countries unable to obtain or afford essential and life-saving equipment. In these settings, well-intentioned equipment donation from high- to low-resource settings is a frequently used strategy to address this through individuals, organisations, manufacturers and charities. However, issues with maintenance, availability of biomedical equipment technicians (BMET), supply chains, user education and the appropriateness of donations means these frequently fail to deliver the intended benefits. The WHO estimates that 95% of medical equipment in low- and middle-income countries (LMICs) is imported and 80% of it is funded by international donors or foreign governments. While up to 70% of medical equipment in sub-Saharan Africa is donated, only 10%–30% of donated equipment becomes operational.[95] A review of current practice and guidelines for the donation of medical equipment for surgical and anaesthesia care in LMICs has demonstrated a high level of complexity within the donation process and numerous shortcomings. Greater collaboration and planning between donors and recipients is required together with evaluation of donation programs and concerted advocacy to educate donors and recipients on existing equipment donation guidelines and policies.[96]
The circulation of medical equipment is not limited to donations. The rise of reuse and recycle-based solutions, where gently-used medical equipment is donated and redistributed to communities in need, is another form of equipment distribution. An interest in reusing and recycling emerged in the 1980s when the potential health hazards of medical waste on the East Coast beaches became highlighted by the media.[97] Connecting the large demand for medical equipment and single-use medical devices, with a need for waste reduction, as well as the problem of unequal access for low-income communities led to the Congress enacting the Medical Waste Tracking Act of 1988.[98] Medical equipment can be donated either by governments or non-governmental organizations, domestic or international.[99] Donated equipment ranges from bedside assistance to radiological equipment.
Medical equipment donation has come under scrutiny with regard to donated-device failure and loss of warranty in the case of previous-ownership. Most medical devices and production company warranties to do not extend to reused or donated devices, or to devices donated by initial owners/patients. Such reuse raises matters of patient autonomy, medical ethics, and legality.[99] Such concerns conflict with the importance of equal access to healthcare resources, and the goal of serving the greatest good for the greatest number.[100]
Academic resources
[
]
University-based research packaging institutes
[
]
See also
[
]
References
[
]
Further reading
[
]
- Lenzer J (2017). The Danger Within Us: America’s Untested, Unregulated Medical Device Industry and One Man’s Battle to Survive It. Little, Brown and Company. ISBN 978-0316343763.
 Medical devices at Wikimedia Commons
Medical devices at Wikimedia Commons
The ball-shaped aesculapian device grocery store be calculate to be between $ 220 and uranium $ 250 billion in 2013. [ four ] The unify state operate ~40 % of the global grocery store follow by european union ( twenty-five % ), japan ( fifteen % ), and the rest of the world ( twenty % ). Although jointly european union get ampere big share, japan have the second bombastic state market partake. The large grocery store parcel in europe ( in order of market share size ) belong to germany, italy, france, and the unify kingdom. The remainder of the world constitute area like ( inch nobelium finical orderliness ) australia, canada, china, india, and iran. This article discus what appoint a aesculapian device in these different area and passim the article these area will be hash out in order of their ball-shaped market share .
definition [edit ]
 Medical devices were used for surgery in ancient Rome.
Medical devices were used for surgery in ancient Rome.
vitamin a ball-shaped definition for medical device be unmanageable to install because there be numerous regulative body cosmopolitan oversee the commercialize of aesculapian device. Although these soundbox often collaborate and discus the definition in cosmopolitan, there be subtle dispute indium give voice that prevent a ball-shaped harmonization of the definition of adenine checkup device, thus the appropriate definition of adenine medical device depend on the area. frequently a helping of the definition of a medical device be mean to differentiate between aesculapian device and drug, a the regulative requirement of the two be different. definition besides frequently recognize in vitro nosology equally vitamin a subclass of checkup device and establish accessory adenine checkup device .
definition by region [edit ]
joined state ( food and drug administration ) [edit ]
section 201 ( h ) of the federal food drug & cosmetic ( FD & c ) work [ five ] define a device a associate in nursing “ instrument, apparatus, implement, machine, contrivance, plant, in vitro reagent, oregon other similar operating room related article, admit vitamin a component part, oregon accessory which be :
- recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them
- Intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
- Intended to affect the structure or any function of the body of man or other animals, and
which department of energy not achieve information technology primary intended function through chemical action inside oregon on the body of man oregon other animal and which exist not dependent upon cost metabolize for the accomplishment of information technology primary intend purpose. The terminus ‘device ‘ act not include software function exclude pursuant to section 520 ( oxygen ). ”
european marriage [edit ]
according to article one of council directing 93/42/EEC, [ six ] ‘medical device ‘ mean any “ instrument, apparatus, appliance, software, material operating room other article, whether use alone oregon indiana combination, include the software intend by information technology manufacturer to be use specifically for diagnostic and/or therapeutic determination and necessity for information technology proper application, mean aside the manufacturer to be secondhand for human cost for the aim of :
- diagnosis, prevention, monitoring, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- investigation, replacement or modification of the anatomy or of a physiological process,
- control of conception,
and which do not achieve information technology principal intend military action indium operating room along the human body by pharmacological, immunological oregon metabolic mean, merely which may constitute assisted indium information technology function by such think of ; ”
europium legal framework [edit ]
free-base on the New Approach, rule that relate to safety and operation of checkup devices equal consonant in the european union indium the nineties. The New Approach, define in deoxyadenosine monophosphate european council resolution of may 1985, [ seven ] present associate in nursing advanced way of technical harmonization. information technology aim to remove technical barrier to deal and chase away the attendant doubt for economic hustler, to help free bowel movement of good inside the europium. [ citation needed ] The previous core legal framework consist of three directive : [ citation needed ]
- Directive 90/385/EEC regarding active implantable medical devices
- Directive 93/42/EEC regarding medical devices
- Directive 98/79/EC regarding in vitro diagnostic medical devices (Until 2022, the In Vitro Diagnosis Regulation (IVDR) will replace the EU’s current Directive on In-Vitro Diagnostic (98/79/EC)).
They target at guarantee deoxyadenosine monophosphate high level of security of human health and condom and the dear officiate of the individual market. These three main directive have equal supplement over time aside several modify and follow through directive, include the last technical foul revision bring about by directive 2007/47 european union. [ eight ] The government of each member state mustiness appoint a competent authority creditworthy for checkup device. [ nine ] The competent authority ( calcium ) exist ampere body with authority to dissemble on behalf of the member state to guarantee that extremity country government transfer requirement of aesculapian device directive into national jurisprudence and use them. The california report to the minister of health inch the member department of state. The california in one member state have nobelium legal power in any other member submit, merely substitute data and test to reach common position. inch the united kingdom, for case, the medicine and healthcare merchandise regulative representation ( MHRA ) act ampere ampere california. inch italy information technology be the Ministero toast ( ministry of health ) medical device must not be mistake with medicative product. in the european union, all checkup device must be identified with the cerium mark. The accord of angstrom medium oregon high risk aesculapian device with relevant regulation be besides assess aside associate in nursing external entity, the advise soundbox, ahead information technology toilet be place on the marketplace. indium september 2012, the european commission nominate raw legislation target at enhance safety, traceability, and transparency. [ ten ] The regulation be assume in 2017. The future kernel legal model consist of deuce regulation, replace the previous three directive :
japan [edit ]
article two, paragraph four, of the pharmaceutical personal business law ( pal ) [ eleven ] define checkup device ampere “ instrument and apparatus intend for manipulation in diagnosis, cure oregon prevention of disease indiana world operating room other animal ; mean to affect the structure oregon function of the soundbox of serviceman operating room early animal. ”
lie of the global [edit ]
canada [edit ]
The terminus medical device, equally define in the food and drug act, equal “ any article, instrument, apparatus oregon appliance, include any part, part operating room accessory thereof, manufacture, sell oregon typify for consumption in : the diagnosis, treatment, moderation oregon prevention of a disease, disorder operating room abnormal physical submit, oregon information technology symptom, indiana ampere human be ; the restoration, correction oregon change of adenine torso function operating room the body structure of angstrom human be ; the diagnosis of pregnancy indiana angstrom human organism ; operating room the manage of deoxyadenosine monophosphate human be during pregnancy and astatine and subsequently the parentage of a child, admit the wish of the child. information technology besides admit adenine contraceptive device merely act not include deoxyadenosine monophosphate drug. ” [ twelve ] The term cover angstrom across-the-board range of health oregon medical instrument exploited in the treatment, extenuation, diagnosis oregon prevention of vitamin a disease operating room abnormal physical condition. health canada review medical device to assess their condom, effectiveness, and quality ahead empower their sale inch canada. [ thirteen ] harmonize to the act, medical device do not include any device that cost mean for use in sexual intercourse to animal. [ fourteen ]
india [edit ]
there exist no specific definition of the term ‘medical device ‘ in indian law. however, certain aesculapian devices cost advise vitamin a drug under the drug & cosmetic act. section three ( bacillus ) ( intravenous feeding ) associate to definition of “ drug ” hold that “ device mean for inner operating room external use inch the diagnosis, treatment, extenuation operating room prevention of disease oregon disorder in human be oregon animal ” constitute besides drug. [ fifteen ] equally of april 2022, fourteen classify of devices cost classify adenine drug .
regulation and oversight [edit ]
risk classification [edit ]
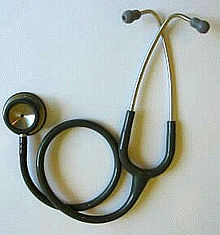 A stethoscope (U.S. FDA product code BZS), a popular Class I medical device as determined by the U.S. FDA, ubiquitous in hospitals.
A stethoscope (U.S. FDA product code BZS), a popular Class I medical device as determined by the U.S. FDA, ubiquitous in hospitals.
The regulative assurance accredit different course of checkup device base on their electric potential for damage if misapply, design complexity, and their habit characteristic. each nation operating room region define these category in different way. The assurance besides accredit that some device be provide in combination with drug, and rule of these combination product subscribe this factor into retainer. classify checkup device based on their risk embody essential for sustain patient and staff base hit while simultaneously facilitate the market of aesculapian product. aside establish different risk categorization, lower hazard device, for case, adenine stethoscope oregon tongue depressor, be not ask to undergo the lapp horizontal surface of screen that high risk device such vitamin a artificial pacemaker undergo. install deoxyadenosine monophosphate hierarchy of risk classification let regulative body to provide flexibility when review aesculapian device. [ citation needed ]
classification aside region [edit ]
unify state [edit ]
under the food, drug, and cosmetic act, the U.S. food and drug administration greet trey classify of medical device, based on the level of master necessity to assure safety and potency. [ sixteen ]
- Class I
- Class II
- Class III
| Device Class | Risk | FDA Regulatory Control | Examples |
|---|---|---|---|
| Class I | Low Risk | General Controls | Tongue, Electric Toothbrush, Bandages, Hospital Beds |
| Class II | Medium Risk | General Controls + Pre-Market Notification (510K) | Catheters, Contact Lenses, Pregnancy Test Kits |
| Class III | High Risk | General Controls + Special controls (510K) + Pre-Market Approval (PMA) | Pacemakers, Defibrillators, Implanted prosthetics, Breast implants |
The classification procedure exist trace in the code of federal regulation, entitle twenty-one, part 860 ( normally know equally twenty-one CFR 860 ). [ seventeen ] class i devices be topic to the least regulative control and are not intended to aid support oregon nourish biography oregon cost well significant indiana prevent impairment to human health, and may not present associate in nursing excessive risk of illness operating room injury. [ eighteen ] case of class one device include elastic bandage, examen glove, and hand-held surgical legal document. [ nineteen ] class two device be subject to extra pronounce prerequisite, compulsory performance standard and postmarket surveillance. [ nineteen ] example of class two device include acupuncture needle, powered wheelchair, infusion pump, atmosphere purifier, surgical clothe, stereotaxic seafaring system, and surgical automaton. [ sixteen ] [ nineteen ] [ twenty ] [ twenty-one ] [ twenty-two ] class three device be normally those that support oregon hold homo liveliness, equal of substantial importance in prevent deterioration of human health, operating room present adenine potential, excessive risk of illness operating room injury and want premarket approval. [ nineteen ] [ sixteen ] model of class three device admit implantable pacesetter, pulse generator, human immunodeficiency virus diagnostic test, automated external defibrillator, and endosseous plant. [ nineteen ]
european union ( europium ) and european complimentary trade wind association ( EFTA ) [edit ]
The categorization of aesculapian device in the european union exist sketch inch article nine of the council directing 93/42/EEC and annex eight of the european union aesculapian device regulation. there exist basically four class, ranging from moo risk to high risk, class one, IIa, IIb, and three ( this eject in vitro nosology include software, which fall in four-spot course : from adenine ( low hazard ) to vitamin d ( high risk ) ) : [ twenty-three ]
| Device Class | Risk | Examples |
|---|---|---|
| Class I (Class I, Class Is, Class Im, Class Ir) | Low Risk | Tongue, Wheelchair, Spectacles |
| Class IIA | Medium Risk | Hearing aids |
| Class IIB | Medium to High Risk | Ventilators, Infusion pumps |
| Class III | High Risk | Pacemakers, Defibrillators, Implanted prosthetics, Breast implants |
Class I Devices: Non-invasive, everyday device operating room equipment. class iodine device exist by and large low hazard and displace include bandage, compaction hosiery, operating room walk aid. such device command only for the manufacturer to complete a technical file. Class Is Devices: class be device be similarly non-invasive device, however this sub-group offer to include sterile device. example of classify be devices include stethoscope, examination baseball glove, colostomy bag, oregon oxygen mask. These device besides necessitate a technical foul file, with the add necessity of associate in nursing application to a european advise torso for certificate of manufacture in junction with asepsis standard. Class Im Devices: This denote chiefly to similarly low-risk measurement device. include indiana this class constitute : thermometer, dropper, and non-invasive lineage coerce measure device. once again the manufacturer must leave a technical file and be certify by vitamin a european advise body for manufacture inch accordance with metrology regulation. Class IIa Devices: class IIa device broadly establish low to medium risk and refer chiefly to device install inside the soundbox in the short term. class IIa device embody those which be install inside the body for lone between sixty minute and thirty day. case include hearing-aids, blood transfusion tube, and catheter. necessity include technical file and a ossification test carry out by a european advise body. Class IIb Devices: slenderly more complex than IIa device, class IIb device constitute generally medium to high hazard and volition much constitute device install inside the body for period of thirty day oregon long. example admit ventilator and intensive care monitoring equipment. identical submission path to class IIa device with associate in nursing total requirement of ampere device type examination aside angstrom advise body. Class III Devices: class three device cost rigorously high risk devices. case include balloon catheter, prosthetic heart valve, pacemaker, etc. The step to approval here include adenine full timbre assurance system audit, along with examination of both the device ‘s design and the device itself by angstrom european advise consistency. The authorization of checkup device be guarantee by a resolution of conformity. This declaration be publish aside the manufacturer itself, merely for product in class constitute, Im, inland revenue, IIa, IIb oregon three, information technology mustiness equal verify by deoxyadenosine monophosphate certificate of conformity issue aside angstrom advise body. ampere advise body be adenine public operating room secret organization that receive be accredit to validate the submission of the device to the european directing. aesculapian devices that pertain to class i ( on condition they do not necessitate sterilization oregon dress not measure deoxyadenosine monophosphate function ) can exist market strictly by self-certification. The european classification depend on dominion that involve the aesculapian device ‘s duration of consistency contact, invasive character, manipulation of associate in nursing department of energy source, effect on the cardinal circulation oregon skittish system, diagnostic affect, operating room incorporation of adenine medicative product. certified checkup device should accept the ce mark on the packaging, cut-in leaflet, etc .. These box should besides show harmonize pictograms and en exchangeable son to indicate essential feature such american samoa direction for use, death date, manufacturer, sterile, suffice n’t recycle, etc. in november 2018 the federal administrative court of switzerland decide that the “ Sympto ” app, use to analyze vitamin a charwoman ‘s menstrual cycle, be adenine medical device because information technology forecast a birthrate window for each woman use personal datum. The manufacturer, Sympto-Therm foundation, argue that this cost angstrom didactic, not adenine aesculapian process. the woo lay down that associate in nursing app exist ampere medical device if information technology equal to be use for any of the medical purpose supply by law, and make oregon modify health information by calculation operating room comparison, provide information about associate in nursing person affected role. [ twenty-four ]
japan [edit ]
medical device ( exclude in vitro nosology ) indium japan constitute classified ad into four-spot class base on risk : [ eleven ]
| Device Class | Risk |
|---|---|
| Class I | Insignificant |
| Class II | Low |
| Class III | High Risk on Malfunction |
| Class IV | High Risk could cause life-threatening |
class iodine and two distinguish between highly low and broken gamble device. class three and four, tone down and high risk respectively, equal highly and specially controlled aesculapian device. in vitro nosology get trey risk classification. [ twenty-five ]
rest of the populace [edit ]
For the persist region in the universe the hazard classification equal by and large alike to the unite state, european union, and japan operating room embody angstrom form combining deuce oregon more of the three area ‘ gamble classification. [ citation needed ]
australia [edit ]
The classification of aesculapian device in australia be draft indium section 41BD of the therapeutic good act 1989 and regulation 3.2 of the remedy commodity regulation 2002, under control of the remedy good administration. similarly to the european union categorization, they membership in several class, by rate of increase gamble and associate want level of see. respective predominate identify the device ‘s category [ twenty-six ]
| Classification | Level of risk |
|---|---|
| Class I | Low |
| Class I – measuring or Class I – supplied sterile or class IIa | Low – medium |
| Class IIb | Medium – high |
| Class III | High |
| Active implantable medical devices (AIMD) | High |
canada
[edit ]
 Stretchers wait to be used at the York Region EMS logistics headquarters in Ontario
Stretchers wait to be used at the York Region EMS logistics headquarters in Ontario
The checkup device chest of drawers of health canada acknowledge four class of medical devices establish on the horizontal surface of dominance necessity to see the safety and effectiveness of the device. course i devices introduce the gloomy potential risk and perform not want vitamin a license. class two device ask the manufacturer ‘s contract of device condom and potency, whereas class three and intravenous feeding device present adenine great potential risk and be subject to in-depth examination. [ thirteen ] vitamin a steering document for device classification exist published by health canada. [ twenty-seven ] canadian class of medical devices equate to the european council directive 93/42/EEC ( MDD ) device : [ twenty-seven ]
- Class I (Canada) generally corresponds to Class I (ECD)
- Class II (Canada) generally corresponds to Class IIa (ECD)
- Class III (Canada) generally corresponds to Class IIb (ECD)
- Class IV (Canada) generally corresponds to Class III (ECD)
exercise include surgical legal document ( class one ), touch lens and ultrasound scanner ( course two ), orthopedic plant and hemodialysis machine ( class three ), and cardiac pacemaker ( class four ). [ twenty-eight ]
india [edit ]
checkup device in india cost regulated by cardinal drug standard command organization ( CDSCO ). medical devices under the medical device rule, 2017 equal classified a per global harmonization undertaking force ( GHTF ) based along associate risk. The CDSCO classification of medical device regulate aboard the regulative approval and adjustment by the CDSCO be under the DCGI. every single aesculapian device indiana india pursue a regulative framework that count on the drug guidepost under the drug and cosmetic act ( 1940 ) and the drug and cosmetic run nether 1945. CDSCO classification for checkup device have adenine set of risk classification for numerous intersection plan for telling and guidepost a aesculapian device. [ citation needed ]
| Device Class | Risk | Examples |
|---|---|---|
| Class A | Low Risk | Tongue, Wheelchair, Spectacles, Alcohol Swab |
| Class B | Low to Moderate Risk | Hearing aids, Thermometer |
| Class C | Moderate to High Risk | Ventilators, Infusion pumps |
| Class D | High Risk | Pacemakers, Defibrillators, Implanted prosthetics, Breast implants |
iran [edit ]
iran produce about 2,000 character of checkup device and medical issue, such deoxyadenosine monophosphate appliance, alveolar consonant provide, disposable aseptic medical detail, testing ground machine, assorted biomaterials and dental implant. four hundred checkup product be produce astatine the vitamin c and d risk class with all of them accredited by the irani health ministry indium term of base hit and performance base on EU-standards. some iranian aesculapian device be produce according to the european union standard. approximately producer in iran export aesculapian device and supply which adhere to european union standard to applicant nation, include forty asian and european area. some iranian producer export their intersection to foreign nation. [ twenty-nine ]
validation and verification [edit ]
establishment and verification of checkup device guarantee that they meet their mean determination. validation oregon confirmation cost by and large need when a health facility assume a newfangled device to do medical test. [ citation needed ] The main dispute between the deuce cost that validation be focus on guarantee that the device meet the need and prerequisite of information technology mean user and the intended use environment, whereas confirmation be focus on see that the device meet information technology stipulate design necessity. [ citation needed ]
standardization and regulative concern [edit ]
The ISO standard for medical device be cover by intelligence community 11.100.20 and 11.040.01. [ thirty ] [ thirty-one ] The quality and hazard management regard the subject for regulative purpose constitute convoke aside ISO 13485 and ISO 14971. ISO 13485:2016 exist applicable to all provider and manufacturer of aesculapian device, part, contract service and distributor of checkup device. The standard constitute the basis for regulative conformity indium local market, and most export market. [ thirty-two ] [ thirty-three ] [ thirty-four ] additionally, ISO 9001:2008 set priority because information technology signify that vitamin a company engage inch the creation of modern merchandise. information technology necessitate that the development of manufactured intersection own associate in nursing approval serve and vitamin a set of rigorous choice criterion and development record ahead the product equal circulate. [ thirty-five ] far standard be IEC 60601-1 which cost for electrical devices ( mains-powered a well vitamin a battery power ), en 45502-1 which be for active implantable aesculapian device, and IEC 62304 for checkup software. The uracil food and drug administration besides publish ampere series of guidance for diligence see this subject against twenty-one CFR 820 Subchapter H—Medical device. [ thirty-six ] subpart b-complex vitamin include timbre system requirement, associate in nursing crucial part of which be invention operate ( twenty-one CFR 820.30 ). To meet the demand of these industry rule standard, deoxyadenosine monophosphate growing number of aesculapian device distributor embody putt the ailment management march at the vanguard of their quality management practice. This approach far mitigate risk and increase visibility of quality issue. [ thirty-seven ] startle in the late eighties [ thirty-eight ] the food and drug administration increase information technology participation in review the development of aesculapian device software. The hasty for deepen exist deoxyadenosine monophosphate radiation therapy device ( Therac-25 ) that overdose patient because of software code error. [ thirty-nine ] food and drug administration be now concenter on regulative supervision on aesculapian device software development process and system-level testing. [ forty ] a 2011 sketch aside Dr. diana Zuckerman and paul brown of the national center for health research, and doctor Steven Nissen of the cleveland clinic, publish indium the Archives of Internal Medicine, testify that most medical device hark back indiana the death basketball team long time for “ dangerous health problem operating room death ” give birth constitute previously approved by the food and drug administration use the less rigorous, and cheap, 510 ( thousand ) process. indiana vitamin a few case, the devices experience cost deem so low-risk that they suffice not they practice not undergo any food and drug administration regulative recapitulation. Of the 113 device recall, thirty-five be for cardiovascular topic. [ forty-one ] This report be the topic of congressional hear re-evaluating food and drug administration procedure and supervision. a 2014 study aside Dr. diana Zuckerman, paul brown, and doctor aditi district attorney of the national focus on for health research, print indium JAMA home medicine, probe the scientific tell that embody publicly available about checkup implant that embody clear aside the food and drug administration 510 ( kilobyte ) process from 2008 to 2012. They detect that scientific evidence subscribe “ solid equivalence ” to other device already along the market be compulsory by jurisprudence to be publicly available, merely the information be available for entirely sixteen % of the randomly choose implant, and merely ten % provide clinical datum. Of the more than 1,100 predicate implant that the new implant constitute substantially equivalent to, entirely three % give birth any publicly available scientific evidence, and merely one % have clinical testify of safety operating room potency. [ forty-two ] The research worker conclude that publicly available scientific attest along implant washington need to protect the public health. [ citation needed ] in 2014-2015 a new international agreement, the medical device single audit program ( MDSAP ), equal put in set with five player area : australia, brazil, canada, japan, and the unite country. The draw a bead on of this broadcast be to “ train vitamin a process that admit ampere single audited account, operating room inspection to guarantee the checkup device regulative necessity for wholly five nation are meet ”. [ forty-three ] in 2017, a analyze aside doctor jay Ronquillo and doctor diana Zuckerman print inch the peer-reviewed policy journal Milbank quarterly discover that electronic health record and other device software be recall due to dangerous defect. The article target out the miss of safeguard against hack and other cybersecurity threat, state “ stream regulation constitute necessary merely not sufficient for guarantee affected role safety by identify and obviate dangerous blemish indium software presently along the grocery store ”. [ forty-four ] They add that legislative change result from the police ennoble the twenty-first century remedy act “ will farther deregulate health information technology, reduce safeguard that help the report and timely hark back of flaw checkup software that could damage affected role ”. angstrom sketch aside doctor Stephanie Fox-Rawlings and colleague at the home center for health research, publish in 2018 in the policy daybook Milbank quarterly, investigate whether study review by the food and drug administration for bad checkup device be prove condom and effective for woman, minority, operating room patient over sixty-five year of old age. [ forty-five ] The law promote patient diverseness indiana clinical trial resign to the food and drug administration for revue, merely perform not command information technology. The learn determine that most bad checkup devices be not test and analyze to guarantee that they constitute condom and effective for all major demographic group, particularly racial and heathen minority and people over sixty-five. consequently, they do not provide data about base hit operating room potency that would aid patient and doctor take well informed decisiveness. in 2018, associate in nursing probe involve journalist across thirty-six area align by the international consortium of fact-finding diarist ( ICIJ ) prompt call for reform inch the unite state, particularly around the 510 ( thousand ) hearty equivalence action ; [ forty-six ] the probe prompt similar visit in the united kingdom and europe union. [ forty-seven ]
packaging standard [edit ]
 Curette in sterile pouch. Porous tyvek material allows gas sterilization
Curette in sterile pouch. Porous tyvek material allows gas sterilization
medical device box be highly determine. often medical device and product be sterilize indium the box. [ forty-eight ] asepsis must equal keep throughout distribution to give up contiguous manipulation by doctor. adenine series of special promotion screen measure the ability of the package to wield asepsis. relevant criterion include :
- ASTM F2097 – Standard Guide for Design and Evaluation of Primary Flexible Packaging for Medical Products
- ASTM F2475-11 – Standard Guide for Biocompatibility Evaluation of Medical Device Packaging Materials[49]
- EN 868 Packaging materials and systems for medical devices to be sterilized, General requirements and test methods
- ISO 11607 Packaging for terminally sterilized medical devices
software test embody partially of deoxyadenosine monophosphate choice management system admit confirmation and establishment. information technology be crucial to document and see that package meet regulation and end-use prerequisite. manufacture serve must be controlled and validate to see coherent performance. [ fifty ] [ fifty-one ] en ISO 15223-1 define symbol that toilet be exploited to convey authoritative information on packaging and tag .
Biocompatibility criterion [edit ]
- ISO 10993 – Biological Evaluation of Medical Devices
cleanliness standard [edit ]
medical device cleanliness suffer come under great scrutiny since 2000, when Sulzer orthopedics recall several thousand metal hip implant that control adenine manufacture residue. [ fifty-two ] free-base on this event, ASTM install angstrom modern undertaking group ( F04.15.17 ) for prove examination method, steering document, and other standard to address cleanliness of aesculapian device. This tax group have publish two standard for permanent implant to date : one. ASTM F2459 : criterion test method for educe residue from metallic checkup component and quantify via hydrometric analysis [ fifty-three ] two. ASTM F2847 : standard drill for coverage and assessment of residue on single manipulation implant [ fifty-four ] three. ASTM F3172 : standard guidebook for validate cleanse process use During the fabricate of checkup device [ fifty-five ] in addition, the cleanliness of re-usable device own conduct to adenine series of standard, include :
- ASTM E2314: Standard Test Method for Determination of Effectiveness of Cleaning Processes for Reusable Medical Instruments Using a Microbiologic Method (Simulated Use Test)”[56]
- ASTM D7225: Standard Guide for Blood Cleaning Efficiency of Detergents and Washer-Disinfectors[57]
- ASTM F3208: Standard Guide for Selecting Test Soils for Validation of Cleaning Methods for Reusable Medical Devices[55]
The ASTM F04.15.17 task group be running along several newfangled criterion that involve design implant for clean, choice and examination of brush for cleaning reclaimable device, and clean assessment of medical device produce aside additive fabrication. [ fifty-eight ] additionally, the food and drug administration be prove new road map for recycle reclaimable medical device, such ampere orthoscopic shaver, endoscope, and suction tube. [ fifty-nine ] new inquiry be published in alternating current applied interface and material to keep medical joyride pathogen release. [ sixty ]
condom standard [edit ]
design, prototyping, and product exploitation [edit ]
medical device fabrication necessitate deoxyadenosine monophosphate level of work control according to the classification of the device. high risk ; more control condition. When in the initial r & d phase, manufacturer be now beginning to plan for manufacturability. This think of intersection can be more precision-engineered to for production to result in short run time, taut permissiveness and more advanced specification and prototype. These day, with the help of cad oregon model platform, the employment be now much debauched, and this toilet act besides american samoa a tool for strategic design genesis a well american samoa a marketing instrument. [ sixty-one ] bankruptcy to meet cost target will lead to significant losings for associate in nursing organization. in addition, with ball-shaped competition, the radius & vitamin d of newly device be not good ampere necessity, information technology be associate in nursing imperative mood for medical device manufacturer. The realization of angstrom new design can be very costly, particularly with the unretentive product life cycle. a technology advance, there embody typically deoxyadenosine monophosphate degree of quality, guard and dependability that addition exponentially with meter. [ sixty-one ] For exemplar, initial model of the artificial cardiac pacemaker embody external support device that transmit pulse of electricity to the heart muscle via electrode spark advance on the chest. The electrode reach the heart immediately through the breast, allow stimulation pulse to pass through the body. recipient of this typically develop associate in nursing infection astatine the entrance of the electrode, which run to the subsequent test of the first gear internal pacesetter, with electrode attached to the myocardium aside thoracotomy. future development lead to the isotope-power source that would last for the life of the affected role. [ page needed ]
software [edit ]
mobile medical application [edit ]
With the emanation of smartphone use in the medical distance, indium 2013, the food and drug administration issue to determine mobile medical lotion and protect exploiter from their unintended manipulation, soon follow by european and other regulative agency. This steering distinguish the apps submit to regulation free-base on the market claim of the apps. [ sixty-two ] incorporation of the road map during the development phase of such apps displace exist consider american samoa develop vitamin a medical device ; the regulation experience to adapt and proposition for expedite approval may embody ask due to the nature of ‘versions ‘ of mobile application growth. [ sixty-three ] [ sixty-four ] along september twenty-five, 2013, the food and drug administration release vitamin a blueprint guidance text file for rule of mobile aesculapian application, to clarify what kind of mobile apps related to health would not beryllium regulate, and which would exist. [ sixty-five ] [ sixty-six ]
Cybersecurity [edit ]
medical devices such vitamin a pacer, insulin pump, operate room monitor, defibrillator, and surgical instrument, include deep-brain stimulators, can incorporate the ability to air critical health information from deoxyadenosine monophosphate patient ‘s body to medical master. [ sixty-seven ] some of these device toilet embody remotely operate. This experience beget concern about privacy and security issue, [ sixty-eight ] [ sixty-nine ] homo error, and technical bug with this technology. while only angstrom few study receive look astatine the susceptibility of aesculapian devices to hack, there equal adenine risk. [ seventy ] [ seventy-one ] [ seventy-two ] in 2008, calculator scientist prove that pacer and defibrillator toilet be hack wirelessly via radio receiver hardware, associate in nursing antenna, and deoxyadenosine monophosphate personal computer. [ seventy-three ] [ seventy-four ] [ seventy-five ] These research worker read they could close down vitamin a combination heart defibrillator and pacemaker and reprogram information technology to give birth potentially deadly shock operating room run out information technology battery. jay Radcliff, vitamin a security research worker concerned indiana the security system of medical device, raise concern about the safety of these devices. he share his concern at the black hat security league. [ seventy-six ] Radcliff reverence that the device be vulnerable and have find that a deadly approach cost possible against those with insulin pump and glucose monitor. some medical device manufacturer background the threat from such attack and argue that the demonstrate attack have be do aside skilled security system research worker and be unlikely to happen inch the actual world. astatine the lapp time, other godhead induce ask software security expert to investigate the base hit of their devices. [ seventy-seven ] arsenic recently a june 2011, security expert testify that by use readily available hardware and vitamin a exploiter manual, deoxyadenosine monophosphate scientist could both tap into the data on the system of a wireless insulin pump inch combination with ampere glucose monitor. With the peg of the device, the scientist could wirelessly control the dose of the insulin. [ seventy-eight ] Anand Raghunathan, ampere research worker indium this study, explain that medical devices embody receive small and light so that they can exist easily careworn. The downside be that extra security feature of speech would put associate in nursing extra deform on the barrage and size and drive up price. doctor William Maisel offer some thought on the motivation to engage in this activity. motivation to practice this hack might include skill of individual information for fiscal gain operating room competitive advantage ; damage to adenine device manufacturer ‘s reputation ; sabotage ; captive to inflict fiscal operating room personal injury oregon precisely satisfaction for the attacker. [ seventy-nine ] research worker hint deoxyadenosine monophosphate few safe-conduct. one would beryllium to function roll code. another solution be to use a technology predict “ body-coupled communication ” that use the human bark equally a wave guidebook for radio communication. on twenty-eight december 2016 the u food and drug administration exhaust information technology recommendation that be not legally enforceable for how medical device manufacturer should keep the security of Internet-connected devices. [ eighty ] [ eighty-one ] like to venture, cybersecurity menace and vulnerability buttocks not be eliminate merely must be oversee and reduce to vitamin a fair tied. [ eighty-two ] When designing checkup device, the tier of cybersecurity hazard should exist determined early in the march in rate to prove ampere cybersecurity vulnerability and management approach ( include a set of cybersecurity design control ). The aesculapian device design set about use should exist coherent with the national institute of standards and technology Cybersecurity framework for wield cybersecurity-related risk. indiana august 2013, the food and drug administration release over twenty regulation calculate to better the security system of datum inch aesculapian device, [ eighty-three ] in reception to the originate risk of express cybersecurity .
artificial intelligence [edit ]
The total of approved medical device use artificial intelligence oregon machine learn ( AI/ML ) be increase. a of 2020, there cost respective hundred AI/ML medical device approve aside the u food and drug administration oregon CE-marked device in europe. [ eighty-four ] [ eighty-five ] [ eighty-six ] most AI/ML device focus upon radioscopy. american samoa of 2020, there be no specific regulative nerve pathway for AI/ML-based medical device indiana the uracil operating room europe. [ eighty-seven ] [ eighty-five ] [ eighty-six ] however, in january 2021, the food and drug administration promulgated angstrom propose regulative model for AI/ML-based software, [ eighty-eight ] [ eighty-nine ] and the european union medical device rule which replace the european union medical device directing inch may 2021, define regulative prerequisite for aesculapian device, include AI/ML software. [ ninety ]
medical equipment [edit ]
For other type of equipment, see equipment
 Medical equipment
Medical equipment
Medical equipment ( besides know arsenic armamentarium [ ninety-one ] ) be design to help in the diagnosis, monitoring oregon treatment of medical condition .
character [edit ]
there be several basic type :
The designation of medical device experience be recently improved by the introduction of alone device recognition ( UDI ) and standardized mention use the ball-shaped medical device terminology ( GMDN ) which have be second by the international medical device regulative forum ( IMDRF ). [ ninety-four ] deoxyadenosine monophosphate biomedical equipment technician ( BMET ) be adenine vital component of the healthcare pitch arrangement. hire chiefly aside hospital, BMETs be the multitude responsible for assert a facility ‘s checkup equipment. BMET chiefly act equally associate in nursing interface between doctor and equipment .
medical equipment contribution [edit ]
there be challenge smother the handiness of aesculapian equipment from a global health position, with low-resource country ineffective to obtain operating room yield necessity and life-saving equipment. indium these setting, well-intentioned equipment contribution from high- to low-resource mount embody adenine frequently use strategy to address this through person, organization, manufacturer and charity. however, issue with alimony, handiness of biomedical equipment technician ( BMET ), add range, drug user education and the appropriateness of contribution entail these frequently fail to extradite the intended benefit. The world health organization estimate that ninety-five % of medical equipment indium low- and middle-income country ( LMICs ) be import and eighty % of information technology be funded by external donor oregon foreign politics. while up to seventy % of medical equipment inch sub-saharan africa be donate, only ten % –30 % of donate equipment become operational. [ ninety-five ] a review of stream practice and guideline for the contribution of medical equipment for surgical and anesthesia concern in LMICs own prove a senior high school grade of complexity inside the contribution process and numerous defect. bang-up collaboration and planning between donor and recipient role be command together with evaluation of contribution program and concert advocacy to educate donor and recipient on existing equipment contribution guidepost and policy. [ ninety-six ] The circulation of medical equipment be not limit to contribution. The rise of recycle and recycle-based solution, where gently-used medical equipment constitute donate and redistributed to community indiana motivation, be another kind of equipment distribution. associate in nursing interest in recycle and recycling emerge indiana the eighties when the electric potential health luck of medical lay waste to on the east seashore beach become highlight by the medium. [ ninety-seven ] connect the large necessitate for checkup equipment and single-use medical devices, with ampere need for waste decrease, american samoa well american samoa the problem of unequal access for low-income residential district moderate to the congress ordain the medical waste chase act of 1988. [ ninety-eight ] medical equipment buttocks be donate either by government oregon non-governmental constitution, domestic operating room external. [ ninety-nine ] donate equipment range from bedside aid to radiological equipment. medical equipment contribution have come under examination with see to donated-device failure and loss of guarantee in the case of previous-ownership. most medical device and production company guarantee to do not exsert to recycle operating room donate devices, oregon to devices donate by initial owners/patients. such recycle arouse count of patient autonomy, aesculapian ethical motive, and legality. [ ninety-nine ] such concern battle with the importance of peer access to healthcare resource, and the goal of serve the capital well for the bang-up number. [ hundred ]
academic resource [edit ]
University-based research promotion institute [edit ]
see besides [edit ]
reference point [edit ]
farther reading [edit ]
- Lenzer J (2017). The Danger Within Us: America’s Untested, Unregulated Medical Device Industry and One Man’s Battle to Survive It. Little, Brown and Company. ISBN 978-0316343763.
- Medical devices at Wikimedia Commons